
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Our Research
We are interested in the synthesis, structure, bonding, magnetism, spectroscopy, and reactivity of organometallic compounds across the periodic table, with a particular emphasis on the chemistry of lanthanide (we include group 3 metals in this definition) and actinide (principally uranium and thorium) complexes which challenge preconceived ideas. The specific topics of our research overlap extensively and we collaborate with others at Manchester, around the UK, and internationally.
Our work divides into five specific themes:
- uranium-metal(loidal) bonds.
- f-block carbenes.
- uranium nitrides.
- f-block single molecule magnets.
- small molecule activation.
Selected examples of our research are outlined below. This is by no means an exhaustive list but we hope it gives a flavour of our research.
f-Block-Metal(loidal) Bonds
Metal-metal bonds are ubiquitous for the d- and p-blocks, and even now known in the s-block, and many groundbreaking advances in our understanding of structure, bonding, and reactivity have emanated from the study of metal-metal bonds. It is somewhat surprising, therefore, to find that until recently f-element-metal bonds were limited to a handful of examples and few of them were structurally characterised. We have set out to remedy this situation by advancing a programme of research to systematically map out the synthesis, structure, and intrinsic reactivity profiles of f-element-metal bonds. Examples of f-element-metal bonds that we have prepared are shown below, Figure 1.
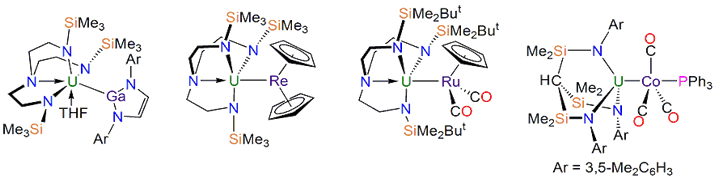
Figure 1. Left to right: the first U-Ga bond; the first U-Re bond; the first U-Ru bond; the first U-Co bond.
This research is already revealing unexpected bonding patterns in uranium-p-block and uranium-d-block metal-metal bonds with the manifestation of, albeit weak, π-bonding in U-Ga and U-Re bonds in addition to the anticipated σ-bonding components. This work is also revealing the differences between 4f and 5f chemical bonding in terms of the extent or absence of covalency in f-element-metal bonds. The issue of covalency in 4f vs 5f bonding is long running and of relevance to the clean up of radioactive waste since ligands which bind covalently can be used to separate uranium from lanthanides and uranium is a reasonable model for plutonium and neptunium which are too 'hot' to handle routinely.
Selected references:
- A Formal High Oxidation State Inverse-Sandwich Diuranium Complex: A New Route to f-Block-Metal Bonds
D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Angew. Chem. Int. Ed., 2011, 50, 10388-10392.
- The Nature of Unsupported Uranium-Ruthenium Bonds: A Combined Experimental and Theoretical Analysis
B. M. Gardner, D. Patel, A. D. Cornish, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Chem. Eur. J., 2011, 17, 11266-11273.
- Synthesis and Structure of [{N(CH2CH2NSiMe3)3}URe(η5-C5H5)2]: A Heterobimetallic Complex with an Unsupported Uranium-Rhenium Bond
B. M. Gardner, J. McMaster, W. Lewis, and S. T. Liddle, Chem. Commun., 2009, 2851-2853.
- σ and π donation in an unsupported uranium-gallium bond
S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones and W. D. Woodul, Angew. Chem. Int. Ed., 2009, 48, 1077-1080.
f-Block Carbenes
f-block carbene chemistry is a relatively underdeveloped area despite the fact carbenes (Fischer, Schrock, N-heterocyclic) are intensively studied and the f-elements are renowned for their ability to perform novel reactions with high reactivities. We have been investigating the synthesis and reactivity of carbenes that posses two phosphorus substituents which can pin the carbene to the lanthanide/actinide centre. Although the group 3/lanthanide carbon bond can be drawn as a double bond we have conclusively showed that it is more accurate to depict it as a dipolar bond, i.e. a latent Y=C double bond akin to Tebbe's reagent. However, the corresponding uranium carbenes can be considered to have a covalent, albeit polarised, U=C double bond. Examples of intrinsically novel reactivity, which are clearly influenced by the level of covalency in these M=C linkages are shown in Scheme 1 below.
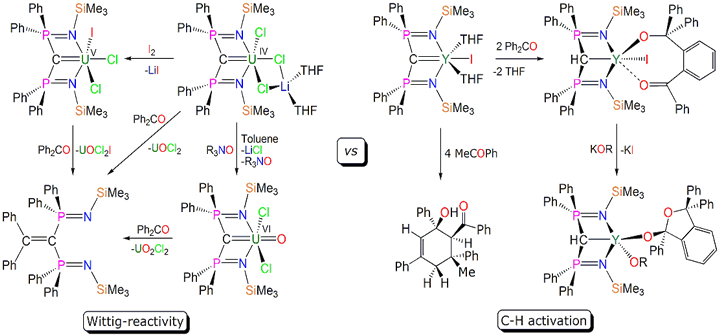
Scheme 1. Reactions of uranium- and yttrium-carbenes.
Reactions which have emerged include: C-H activation and C-C bond formation with benzophenone; 1,2-migratory insertion chemistry; organocatalytic reactivity of benzophenone producing dimerisation of the carbene; [2+2] cycloaddition reactions; carbo-potassiation of the Y=C bond; hydro-alkoxylation of the Y=C bond; conventional salt elimination derivatisation chemistry generating alkyl, amide, and metal complexes. We are currently investigating other reactivity patterns and the potential for these systems to form the basis of novel catalytic cycles for C-C and C-N bond forming reactions.
Selected references:
- Synthesis, Characterization, and Reactivity of a Uranium(VI) Carbene Imido Oxo Complex
E. Lu, O. J. Cooper, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake, and S. T. Liddle, Angew. Chem. Int. Ed., 2014, 53, 6696-6700.
- Synthesis of a Uranium(VI)-Carbene: Reductive Formation of Uranyl(V)-Methanides, Oxidative Preparation of a [R2C=U=O]2+ Analogue of the [O=U=O]2+ Uranyl Ion (R = Ph2PNSiMe3), and Comparison of the Nature of UIV=C, UV=C and UVI=C Double Bonds
D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake, and S. T. Liddle, J. Am. Chem. Soc., 2012, 134, 10047-10054.
- Uranium-Carbon Multiple Bonding: Facile Access to the Pentavalent Uranium Carbene [U{C(PPh2NSiMe3)2}(Cl)2(I)] and Comparison of UV=C and UIV=C Double Bonds
O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake, and S. T. Liddle, Angew. Chem. Int. Ed. 2011, 50, 2383-2386.
- Regioselective C-H Activation and Sequential C-C and C-O Bond Formation Reactions of Aryl Ketones Promoted by an Yttrium Carbene
D. P. Mills, L. Soutar, W. Lewis, A. J. Blake, and S. T. Liddle, J. Am. Chem. Soc., 2010, 132, 14379-14381.
Uranium Nitrides
For many years there has been intense interest in isolating a terminal uranium-nitrogen triple bond. Previously, this linkage had only been observed in matrix isolation experiments at ca 10 K and all molecular uranium nitrides contained bridging nitride atoms or were fleeting intermediates under photochemical conditions. The linkage was long sought after because of the ongoing debate regarding the nature (5f vs 6d orbital participation) and extent of covalency in uranium-ligand multiple bond linkages. Using a very bulky ligand (colloquially referred to as trenormous in the group) combined with a mild oxidation strategy using sodium azide, followed by gentle removal of the sodium ion with 12-crown-4 ether, we have isolated the uranium-nitride triple bond linkage, Scheme 2.
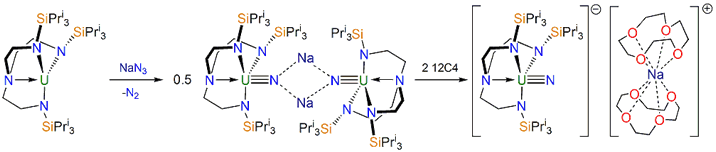
Scheme 2. Synthesis of a terminal uranium-nitride triple bond linkage.
The terminal uranium-nitride complex is an excellent example of the interdisciplinary research that is required in this area of chemistry. The nitride complex was characterised by X-ray crystallography, 1H NMR spectroscopy, FTIR spectroscopy, 15N-isotopomer labelling, EPR spectroscopy, CHN microanalysis, UV/Vis/NIR spectroscopy, SQUID magnetometry, Evans method solution magnetic moment, DFT calculations, reaction with water to produce and trap ammonia, and reaction with Me3SiCl to produce an imido derivative. This nitride can be oxidised to afford a uranium(VI)-nitride and offers the possibility for reduction chemistry as well as metathesis and small molecule activation, for example reductive carbonylation by carbon monoxide. This work has also set the scene for other novel uranium-ligand multiple bonds such as terminal parent U=NH imido, U=PH phosphinidene, and U=AsH arsinidene linkages, and even a uranium-arsenido triple bond linkage.
Selected references:
- Triamidoamine Uranium(IV)-Arsenic Complexes Containing One-, Two-, and Three-fold U-As Bonding Interactions
B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Nat. Chem., 2015, 7, 582-590.
- Synthesis and Characterization of an f-Block Terminal Parent Imido [U=NH] Complex: A Masked Uranium(IV)-Nitride
D. M. King, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake, and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619-5622.
- Triamidoamine-Uranium(IV)-Stabilized Terminal Parent Phosphide and Phosphinidene Complexes
B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Angew. Chem. Int. Ed., 2014, 53, 4484-4488.
- Two-Electron Reductive Carbonylation of Terminal Uranium(V) and Uranium(VI) Nitrides to Cyanate by Carbon Monoxide
P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Angew. Chem. Int. Ed., 2014, 53, 10412-10415.
- Isolation and characterisation of a uranium(VI)-nitride triple bond
D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Nat. Chem., 2013, 5, 482-488.
- Synthesis and Structure of a Terminal Uranium Nitride Complex
D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Science, 2012, 337, 717-720.
f-Block Single Molecule Magnet Systems
Compounds which exhibit magnetic properties that derive from molecular rather than bulk properties are called single molecule magnets (SMMs). There is intense interest in this area because of the potential applications in ultra-high density data storage, spintronics, and quantum computing. The f-block elements hold significant promise in this regard because of their inherently high magnetic anisotropies and large spin-orbit coupling.
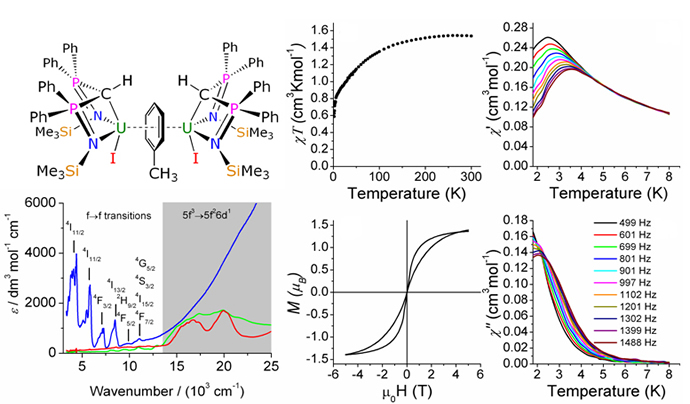
Figure 2. Top left: Inverted sandwich arene-bridged diuranium complex. Bottom left: UV/Vis/NIR of the SMM (blue trace) with two uranium(III) complexes (red and green traces) for comparison. Right: Magnetic data showing hysteresis and slow relaxation in the in- and out-of-phase AC-susceptibility measurements which are hallmarks of single molecule magnetism.
We have recently discovered that an inverted sandwich, arene-bridged diuranium complex displays single molecule magnetism. The nature of the magnetic coupling between the two uranium centres is still unclear, but it would seem that the arene bridge, with a highly anisotropic coordination environment, favours SMM behaviour, Figure 2. More recently, we have established that single-ion uranium(V) in a strongly linear and axial coordination geometry supported by terminal oxo and triamidoamine ligand sets can also exhibit SMM behaviour.
Selected references:
- The Inherent Single Molecule Magnet Character of Trivalent Uranium
F. Moro, D. P. Mills, S. T. Liddle, and J. van Slageren, Angew. Chem. Int. Ed., 2013, 52, 3430-3433.
- Single-Molecule-Magnetism in a Single-Ion Triamidoamine Uranium(V) Terminal Mono-Oxo Complex
D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes, and S. T. Liddle, Angew. Chem. Int. Ed., 2013, 52, 4921-4924.
- A delocalised arene-bridged diuranium single molecule magnet
D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake, and S. T. Liddle, Nature Chem., 2011, 3, 454-460.
Small Molecule Activation
Carbon monoxide (CO) is readily available and in principle an excellent resource from which to produce industrial hydrocarbon feedstocks as alternatives to crude oil. The concept of directly homologating CO is very appealing, but the triple bond in CO is very strong, with a bond energy of ~257 kcal mol–1, and direct dimerisation of CO to O=C=C=O is highly unfavourable with ΔGº 298K ≈ +73 kcal mol–1. Although direct 1,1-migratory insertion of CO into an organometallic M-R bond to give M-C(O)R is a favourable process for many metals, and a key step in industrially important C-C bond formation reactions, e.g. hydroformylation, double insertion to give M-C(O)C(O)R is usually energetically unfeasible. However, combining the coupling of CO with reduction results in a thermodynamically feasible process to give a family of oxocarbon dianions CnOn2– (n = 2-6), including the reductively dimerised ethyne diolate, –O-C≡C-O–, as building blocks for more complex and value-added organic molecules. Thus, the f-block metals with their high reduction potentials represent an attractive approach to homologating CO, but CO has proven remarkably resistant to selective homologation and the few complexes that can effect this transformation cannot be recycled because liberation of the homologated product destroys the complexes or they are substitutionally inert.
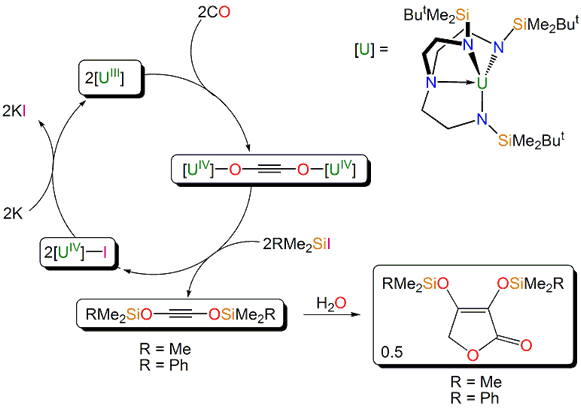
Scheme 3. A synthetic cycle for the reductive homologation of carbon monoxide.
By employing a sterically demanding, rigid, and preorganised Tren-uranium complex we can effect reductive homologation of CO, Scheme 3. Importantly, this complex is ammenable to liberation of the coupled product. Thus, we can close the synthetic cycle and the liberated product reacts further to ultimately yield substituted furanones. Overall, this process combines CO and silyl halides to produce furanones using a recyclable uranium promoter.
Selected references:
- Homologation and Functionalization of Carbon Monoxide by a Recyclable Uranium Complex
B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake, and S. T. Liddle, Proc. Nat. Acad. Sci. USA, 2012, 109, 9265-9270.