This site is under construction...
|
We use molecular simulation and coarse grained techniques to obtain thermodynamic and transport properties of systems that are relevant for the chemical industry. In particular, we are interested in systems containing surfactants and liquid crystals, as well as fluids in confined systems, like gases and liquids in porous carbons, zeolites or related mesoporous materials. Currently we are working on the following projects: Hybrid porous materials (DeSANNS, FP6) Dynamics of formation of porous materials (DeSANNS, FP6)
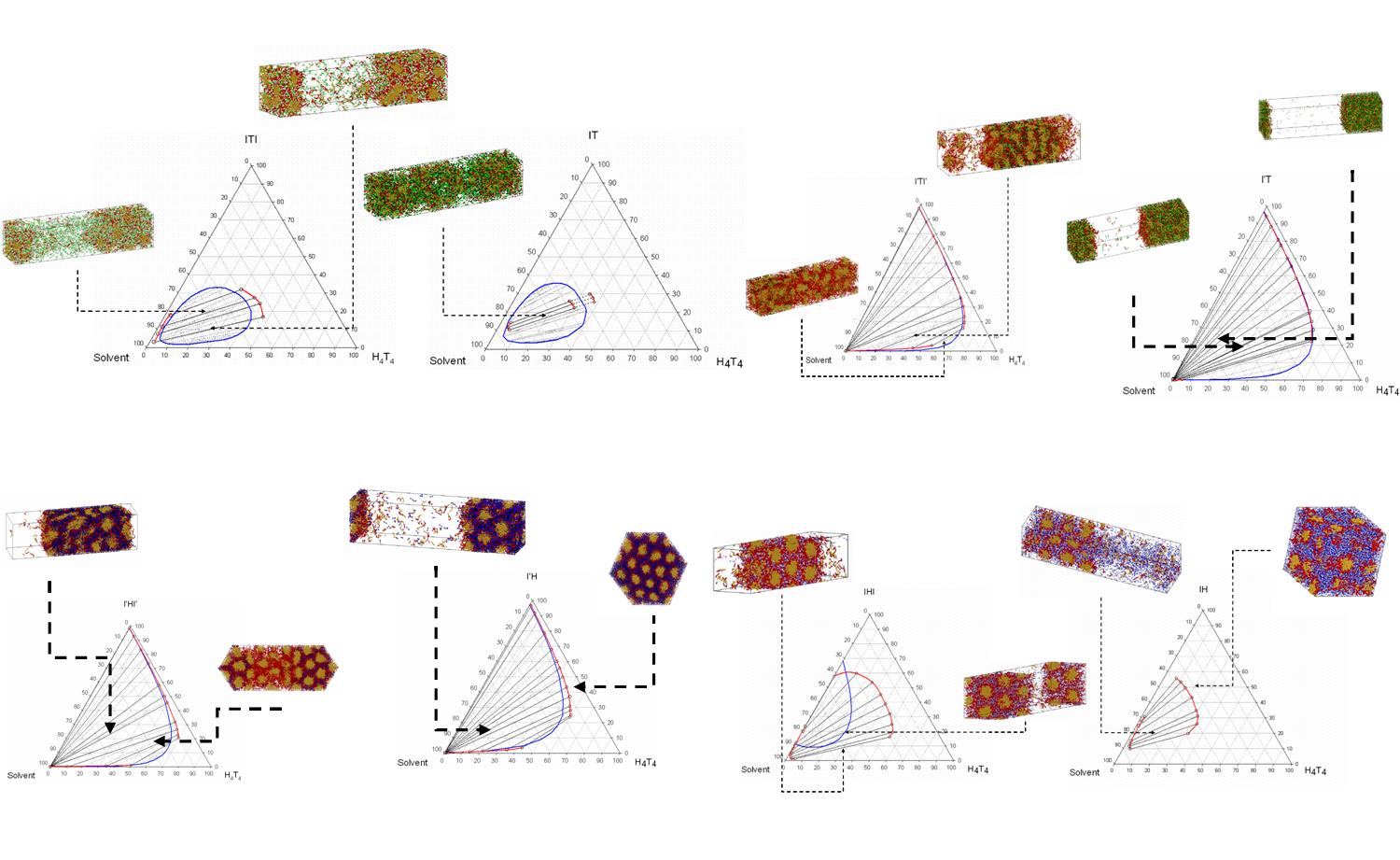
Surfactant-self assembly and the formation of porous materials (DeSANNS, FP6) We are interested in understanding the formation of mesoporous solids where surfactant self assemblies are used as templates to direct the structure of the material. Such materials were first reported in the early 1990's by researchers in Mobil, and soon after several synthesis methods were reported using a variety of cationic, anionic, non-ionic surfactants or every block copolymers, to control the pore size and shape of the material. Preliminary result allow us to understand the limitations for formation of ordered materials with hybrid precursors. Our current interest is to abandon the assumption that these systems are at equilibrium and study the effect of the extent of the silica condensation reaction upon the formation of ordered structures.
Behaviour of surfactant self-assemblies Solubilization and release of solutes from micelles has applications in many fields, including drug delivery, release of agrochemicals, personal care and other formulated products. We are using Dissipative Particle Dynamics to study the effect of the solvent in solutions containing triblock type surfactants. In particular we are interested in understanding the dynamics of the micelles when the solvent properties are suddenly changed.
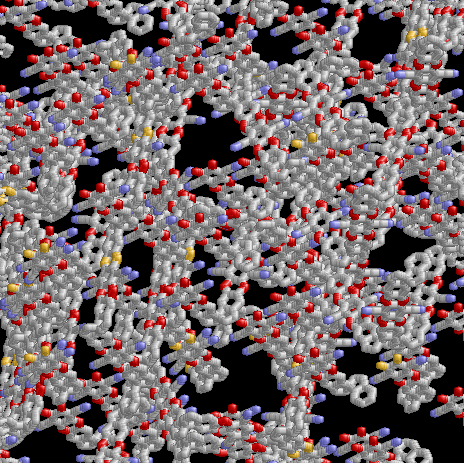
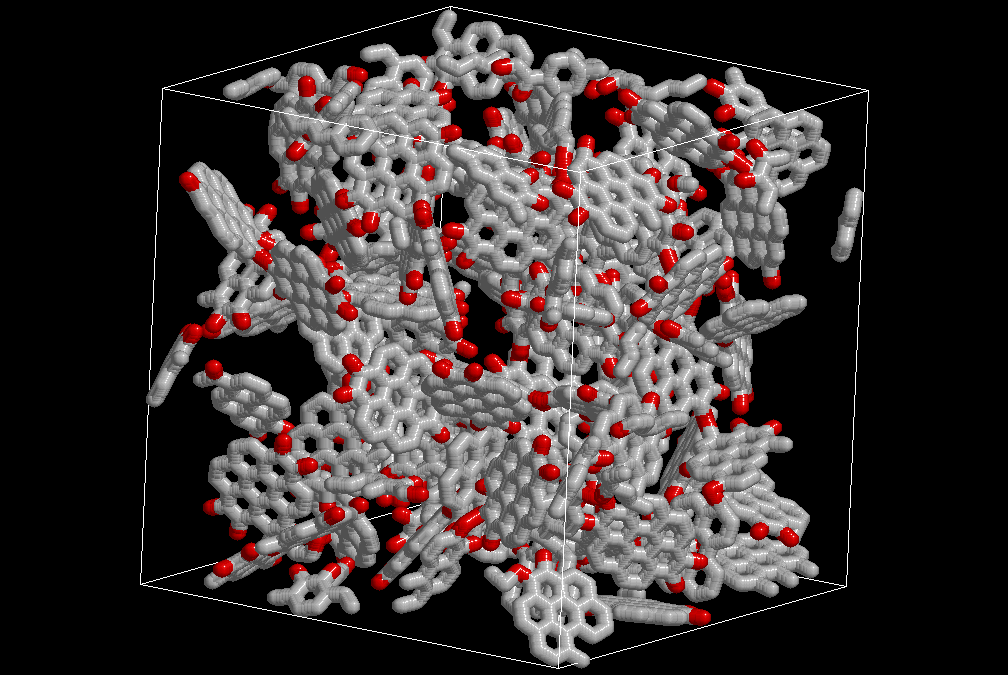
Modelling Polymers of Intrinsic Microporosity in collaboration with P. Budd, Chemistry (U. Manchester), Coray Colina and Greg Larsen from Materials Science (Penn State)
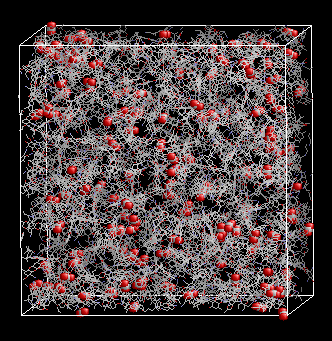
Characterization of activated carbons Activated carbons are probably the most widely used adsorbents, from industrial gas storage, to prevention of poison absorption by the gastrointestinal tract. Activated carbons are disordered materials, composed mainly of graphite platelets that can be stacked in different orientations. The distance between the platelets (pore size distribution), the presence of functional groups on the edges of these platelets (hydroxyl, carbonyl or carboxyl groups), and their distribution define their applications. Due to the high surface area of activated carbons, adsorption of different probe molecules is among the most important techniques used for its characterization. Unfortunately, interpretation of the experimentally measured data often requires the use of models that make important approximations (like the assumption of infinite slit pores) that may result in unrealistic descriptions of the material. More realistic descriptions of disordered carbonaceous materials have been recently obtained using molecular simulations [see for example, Pikunic, et al. Langmuir 2003, 19, 8586; Biggs, et al. Langmuir 2004, 20, 5786; Biggs and Buts Mol. Sim. 2006, 32, 579.].
Adsorption and transport in microporous materials (in collaboration with the IFP)
|
The content in these pages reflect only the author's view. The University of Manchester or the funding agencies are not liable for the content or any use that may be made of the information contained in these pages.