Department of Chemical Engineering
Dr Thomas Rodgers
Department of Chemical Engineering
Dr Thomas Rodgers
Step Two: Defining the molecules and their Interactions - FIELD
|
First, you need to create an input file that will contain information about the molecules and their interactions. The system you will build contains diblock amphiphilic molecules in solvent, in 55% concentration. Each molecule consists of two beads – a hydrophilic one (type A) and a hydrophobic one (type B). There are two ways to create the molecule. One way is to create it manually and the second way is to use the DL_MESO built-in utility molecule.exe. In this exercise, you will make use of the molecule.exe tool. To launch the tool, type: ~/dl_meso_2.6/BIN/molecule.exe The following figure shows the questions you will be asked and what you should enter to build the AB dimer. Analytically, you will be prompted to enter the number of species that will compose the molecule, their names and the number of different molecules you want to design. In this case, we have 2 species of beads, named A and B and 1 type of molecule. Next, you will be prompted to define the intra-molecular interactions, i.e. the bonds, angles and dihedrals. Since the molecule consists only of 2 beads, you only have 1 bond and no angles or dihedrals. Enter 1 as the number of bond types and select 1 for the bond potential, which corresponds to the harmonic potential. The spring force constant and the equilibrium bond length should be 100.0 and 0.50, respectively. Now, the program will start designing the molecule. Give your molecule the name ‘AB’ and define the molecule population as 3300. Do not permit isomers (False - F). Define the cube size as 2 and the bond length as 0.50. After this, you will be asked how many chains you have as well as how many beads per chain. Here, you have 1 chain and 2 beads/chain. The type A can be defined as species 1 and type B as species 2. Finally, the default species for the beads in the molecule will be requested: select A and then enter 1 as the bead number for each of the other species (i.e. B). By typing 0 you terminate the utility. If you make a mistake, type Ctrl+C, delete the FIELD file and start from scratch. 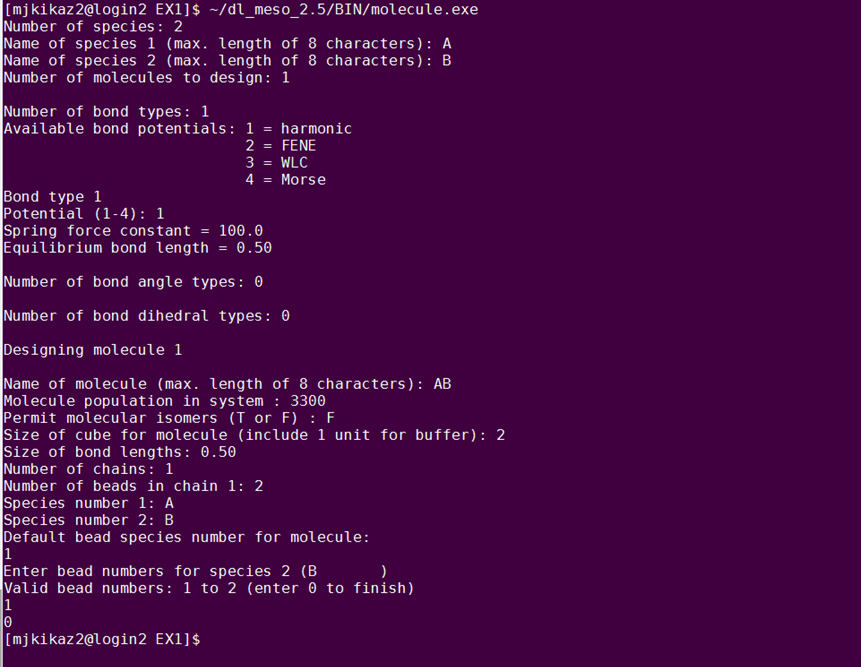
You should have now a FIELD file that contains all the above information and looks like that: 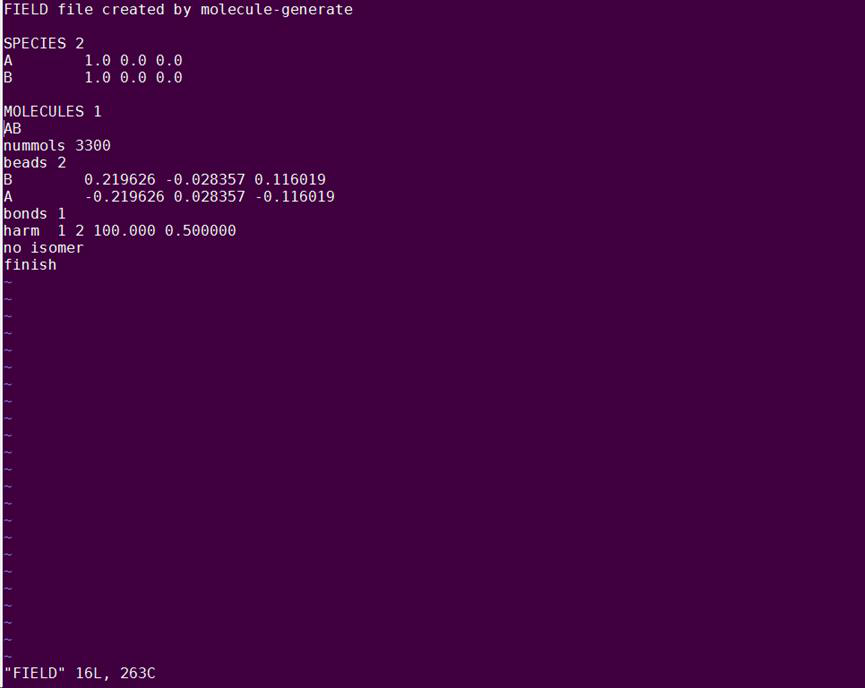
You have built the dimer, but you also need to add the solvent and describe the intermolecular interactions between the beads. So next, you will edit the FIELD file to include the missing information: nano FIELD The first line is a description of the simulation. You can change that into: DL_MESO Amphiphile Mesophase (55% composition) Change the number of species to 3 to account for the solvent (S). Add a line below the description of species A and B, for the solvent S, which has mass equal to 1, charge equal to zero and a population of 5400 beads. At the bottom of the file, you should include the non-bonded interactions for the DPD particles. First, add a line that starts with the keyword INTERACTIONS and define the number of interactions as 6: this number corresponds to all the possible pairs of beads (AA, AB, AS, BB, BS, SS). Write these pairs in a column and add the dpd interaction potential in the second column, as in the following figure. Define the repulsion parameters (aij) for each pair, as below. Remember that a higher value of aij corresponds to higher repulsion. The cut-off distance is always equal to unity and the friction coefficient γ is set equal to 5.625. The bottom line of the file should contain the word CLOSE, which closes the file. Your FIELD file should look like in the figure below. The lines starting with ‘#’ are comments and must be omitted. Finally, save and close the file. 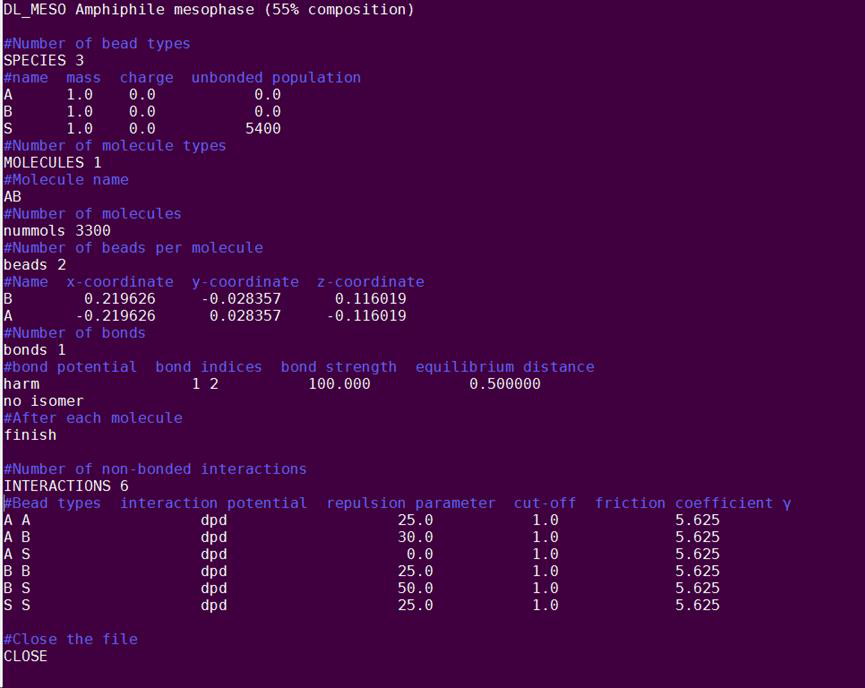
|