Dr Neil A. Burton
Computational and Theoretical Chemistry Group
School of Chemistry
University of Manchester
Manchester, M13 9PL
Email: neil.burton@manchester.ac.uk
Dr Neil A. BurtonComputational and Theoretical Chemistry Group School of Chemistry University of Manchester Manchester, M13 9PL Email: neil.burton@manchester.ac.uk |
Computational chemistry - the study of chemical processes and properties using computational models and advanced computing technology. Computational chemistry is a practical branch of theoretical chemistry, with computer programs now widely employed to study a variety of biological, environmental, solid-state and chemical processes involving one to many thousands of atoms or molecules. Quite often the term molecular modelling is used within industry or the drug discovery process, emphasizing the study of molecular structure and molecular interactions.
Computer simulations of the dynamics (molecular dynamics) and electronic structures (quantum chemistry) of molecular systems and chemical reactions allows us to directly visualize and understand complex chemical processes at an atomic level of detail, and provides a valuable complement to experimental study, which is often limited to interpretation of measured data or the study of only stable chemical structures.
[Click here for information on MChem Projects]
Our research focuses on the development, implementation and application of computational methods to simulate chemical reactivity in the condensed phase, particularly to understand catalysis and chemical mechanisms. Some recent highlights include (click on the pictures for relevant publications):
Modelling techniques can be used to explore the structures and transition states of biochemical reactions which may ultimately impact the pharmaceutical drug discovery process.
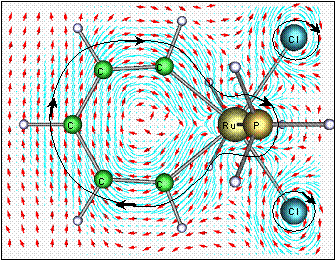
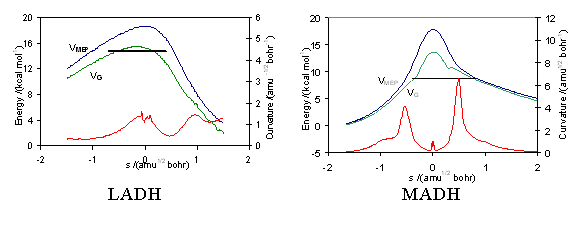
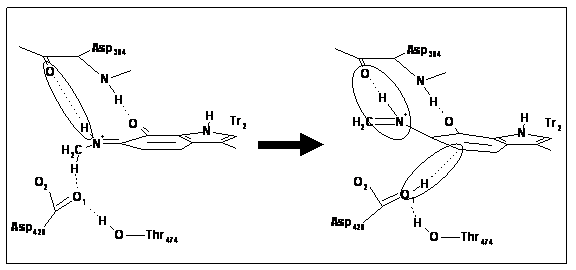
A topical question in biology in the last decade has been the role and importance of protein dynamics and quantum mechanics in the catalytic mechanisms of common enzyme reactions. Chemical reactions which involve the transfer of protons are amongst the most abundant and the most important, particularly when breaking C-H bonds since these can be particularly high energy and slow processes. In some classes of enzyme unusual experimental kinetic behaviour has been observed consistent with a hypothesis that quantum mechanical tunnelling effects may play a significant role to reduce the classical barriers in these reactions. For the enzyme methylamine dehydrogenase, QM/MM methods have been combined with direct dynamics calculations to obtain accurate reaction profiles for the proton transfer step of the reaction (see below). Variational transition state theory and semi-classical tunnelling corrections have been applied to compute the potential energy surfaces, rates of reaction, and the high primary deuterium kinetic isotope effects in order to complement experimental measurements and to understand the mechanism. Quantum mechanical tunnelling is indeed significant and is strongly mediated by the protein. |
 |
 |
 |
|---|
Computational chemistry methods allow us to predict and study transition state structures and reaction mechanisms, but the size and complexity of most real chemical systems, particularly in condensed phases, makes high level ab initio quantum mechanical calculations impractical, even on modern computers. Thus there is a need to develop and augment the way in which we use theoretical techniques in order to more realistically describe the chemistry. In our research we often use theoretical models of the condensed phase environment and model the atoms of a protein or solvent at a more approximate theoretical level, treating the most important regions where bonds are forming and breaking using quantum mechanical methods. In particular, we have developed a range of hybrid quantum mechanical and molecular mechanical (QM/MM) simulation approaches in order to accurately model chemical reactions involving thousands of atoms. A key component of this research is the use of efficient semi-empirical quantum chemistry methods to describe the molecules using specific reaction parameters which reproduce the accurate energetics and structures, including those involving transition metals, obtained from high level model calculations. This has allowed us to use simulation methods, such as molecular dynamics, to obtain reaction or activation free energies or potentials of mean force along a reaction coordinate using quantum potentials, thus accounting for entropy and the many configurations which the molecules can adopt.
 |
Protein and solvent environments can be particularly influential in mediating chemical reactions and properties, either acting as a framework to promote catalysis or to mediate the electronic properties of cofactors and substrates. By studying smaller chemical models, and combining the benefits of experiment and computation, we may obtain new insight into reaction mechanisms at an atomic level and predict kinetic behaviour of large complex macromolecular systems. |
 |
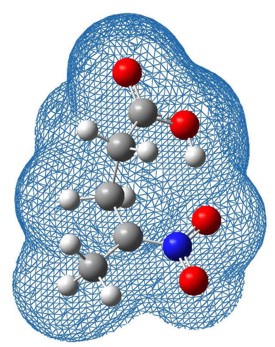
For example, in collaboration with experimental colleagues in physical organic chemistry (Dr Ian Watt), we have been studying the reactions of proton transfer reactions in intramolecular complexes, which are models or mimics for enzyme reactions, in order to understand the detailed mechanisms taking place. In these studies of nitroalkane complexes, tunnelling only plays a minor role in the mechanism but solvent relaxation is extremely important. |
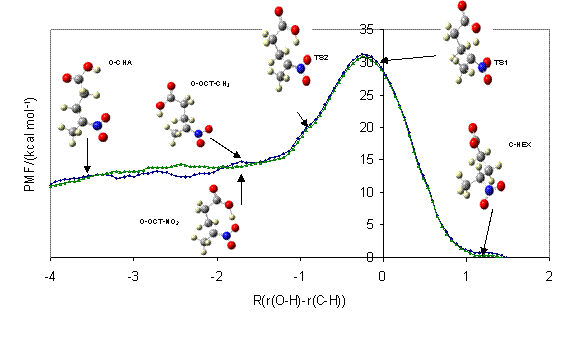 |
|---|
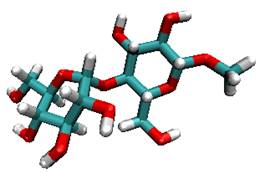 |
Molecules can assume a great many conformations, and chemical structures are dynamic entities. One of the main challenges of computational chemistry is to account for the many possible structures and reaction pathways which are encountered in real chemical reactions. We have recently applied novel sampling methods such as transition path sampling to study the conformational free-energy landscape and rates of molecular conformational change in flexible carbohydrate molecules. |
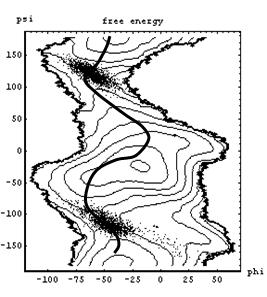 |
|---|
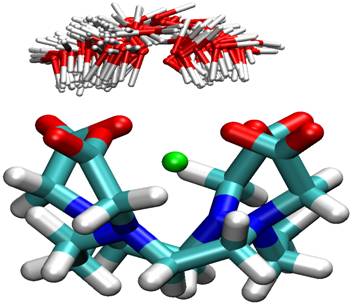 |
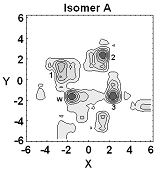
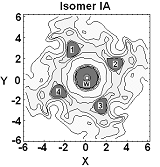
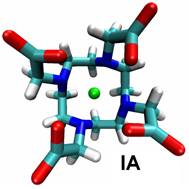
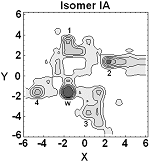
Transition path sampling, which involves the analysis of many thousands of reactive pathways, was also employed to understand the role of solvent in the ligand exchange reaction of lanthanide complexes, an important process in MRI imaging. An analysis of thousands of transition states in the dissociative ligand exchange of water by Gd-DOTA shows that the solvation shell is largely responsible for the increased rate of exchange observed in the IA isomer. Below, the left map of each isomer shows the probability density of water binding in the reactant and the right maps show the water distribution in the transition states. |
|---|
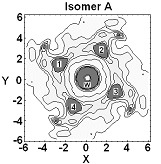 |
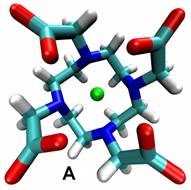 |
 |
 |
 |
 |
|---|
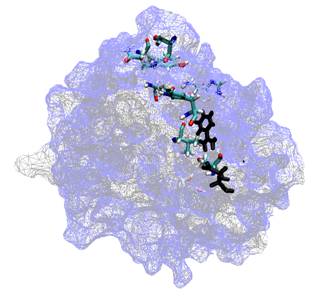
Ligand binding processes are also particularly important in the drug discovery process and much of our research effort is directed to inform this process. Computatational chemistry methods can be particularly useful to understand the mechanisms and trends of potential inhibitors, offering increased accuracy over empirical force-field or molecular mechanics modelling approaches or the scoring functions employed for virtual screening. QM/MM methods are particularly effective when inhibition involves covalent bonding or where the interactions will be poorly described by traditional classical potentials. For example, we have used QM/MM methods to understand the binding trends of some peptidyl-alpha-ketoheterocyclic inhibitors of elastase (below left) explaining the the trend of the binding efficacy of the inhibitors, and that the two best operate by a covalent mechanism (below right).
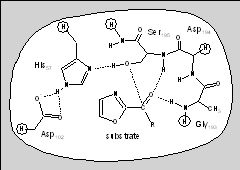 |
 |
|---|
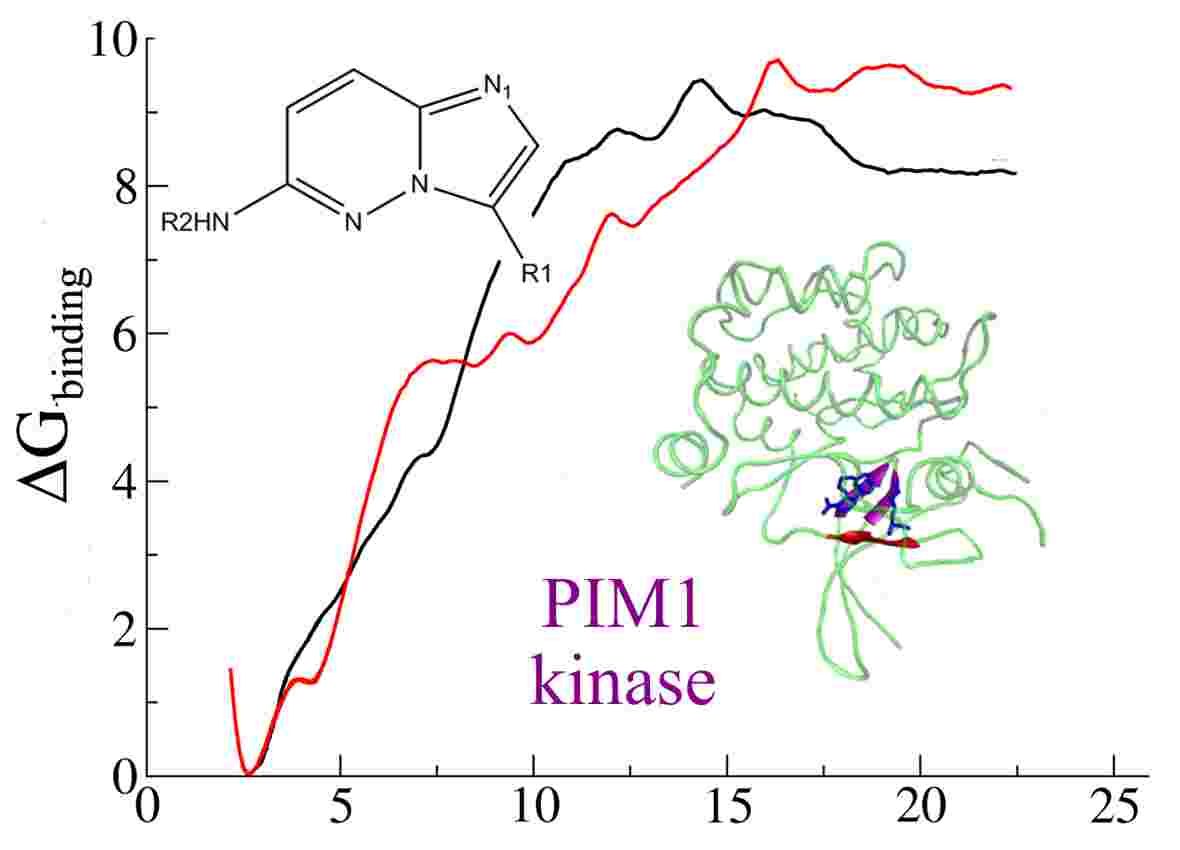
In more recent research we have employed empirical corrections to describe the weak hydrophobic dispersion forces absent in conventional density functional methods in order to more accurately describe binding events. These, together with advanced simulation methods, particularly including umbrella sampling and our hybrid QM/MM approaches, are currently being used to study some topical protein-ligand binding problems and to determine the Gibbs energies of binding to understand and better predict drug efficacy. For example, protease (such as HIV-1RT) and kinase enzymes (such as PIM1) are important targets for inhibition and are of great interest to the pharmaceutical industry (see below). |
 |
|---|
 |
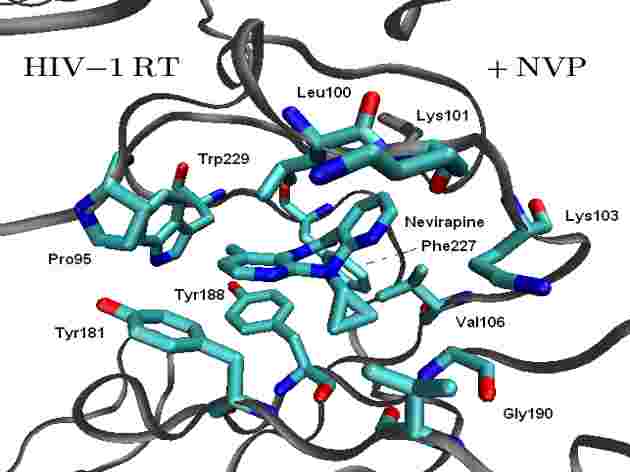 |
|---|
 |
Electronic structures, particularly of transition metal complexes can be difficult to understand and as a result they can react in quite complex and varied reactions. Quantum chemistry, and in particular density functional theory (DFT) is a particularly valuable method to understand the mechanisms of metalloenzymes which are particularly important catalysts. Two recent examples, with importance for future sustainable energy production and synthesis are the hydrogenase and halogenase enzymes respectively. |
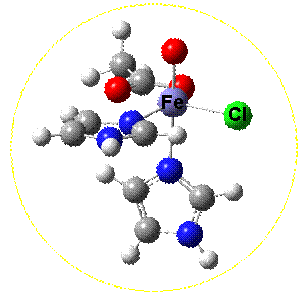 |
|---|
An important aspect of computational quantum chemistry is the ability to explain the structure and bonding of quite unusual electronic structures. Aromaticity is an important concept in chemistry, and is used to explain the thermodynamic and reactive stability of a wide range of molecules, the most common being benzene. Magnetic properties, such as NMR shielding, are useful indicators of electron density distributions in molecules and computational study of the magnetically induced currents in molecules can be a useful indicator of electron delocalisation and bonding. We have extended these approaches to study metal containing molecules, such as the metallabenzenes and changes in aromaticity can be understood in terms of electronic perturbations. For example we have used these criteria to explain why the 18e metallabenzenes (MC5 rings) of Ir and Rh are aromatic and the corresponding 16e complexes of Ru and Os are anti-aromatic. These metallo-complexes and the chelate rings are important in some metalloenzyme reactions. |
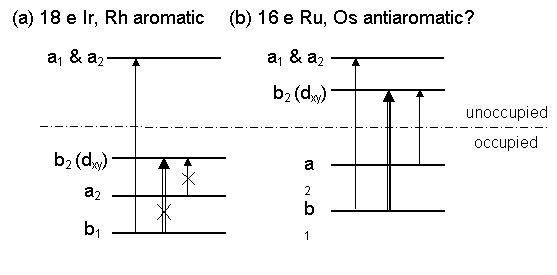 |
|---|
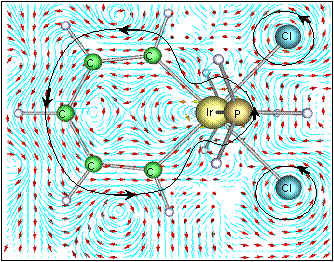 |
|
|---|---|
18e aromatic? |
16e anti-aromatic? |
|
Much of our ongoing research is currently directed towards the modelling of actinide chemistry, particularly the speciation and interaction of important complexes, which are relevant to the nuclear industry, with common minerals such as calcite. Exploration of the chemistry of the actinide elements, such as Uranium, Neptunium and Plutonium, is especially important to comprehend the long term behaviour of nuclear wastes in repository storage facilities and the transport properties of radioactive species in the environment. Research in this theme is being undertaken in collaboration with Professors Livens (Radiochemistry) and Vaughan (Environmental Sciences) . We are also active members of the Nuclear FiRST DTC (Manchester and Sheffield). There are many advantages to investigate actinide chemistry through atomic and nano-scale computational modelling: • no radiation or safety issues to worry about so that we can complement experimental observations and analysis and then make predictions prohibited experimentally;
• can directly study fundamental chemical processes at the atomic level. In actinide chemistry the electronic structure, and especially the role of the f-electrons, is often crucial to accurately predict reactivity and molecular structure.
| 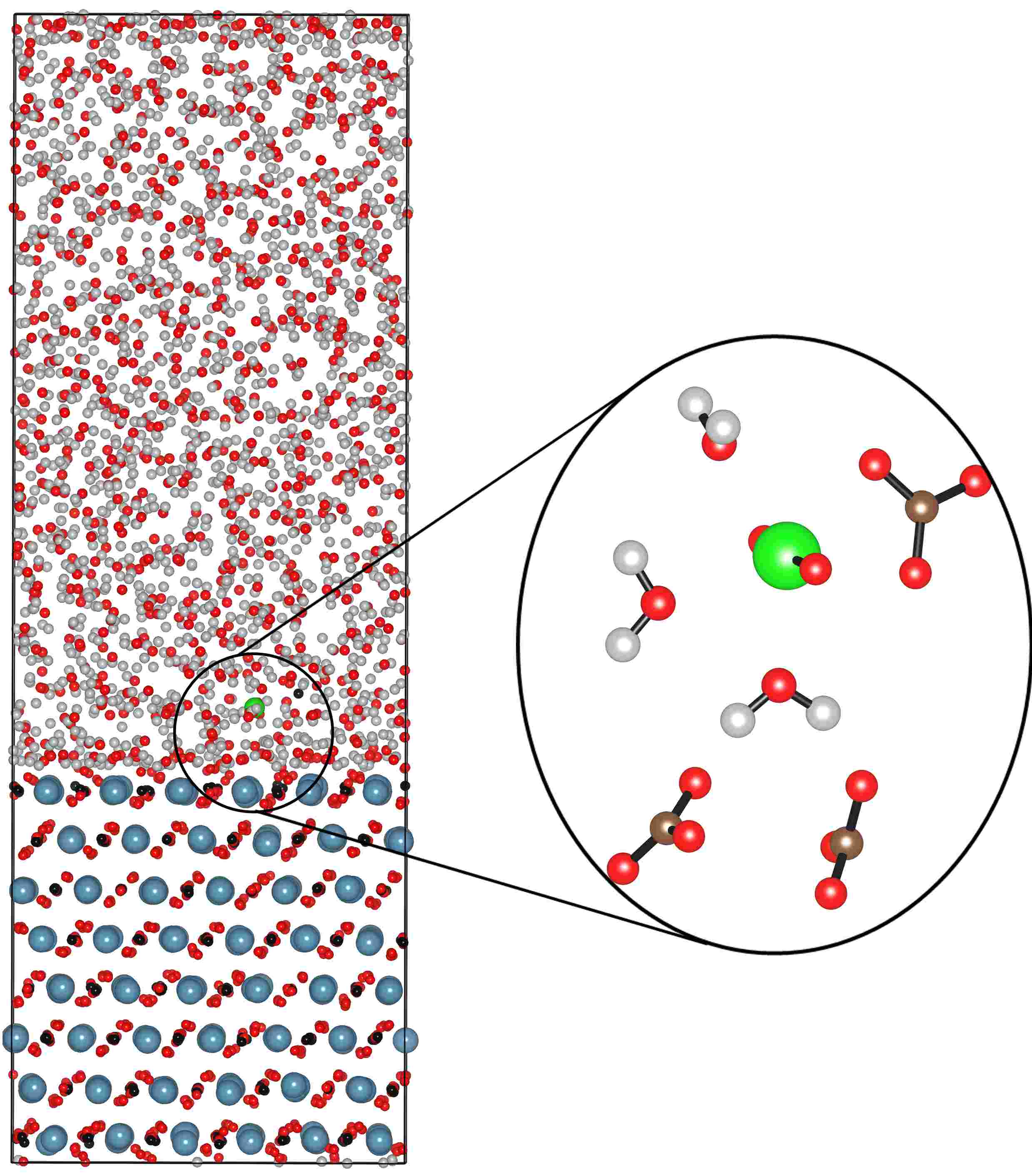 |
We are also developing our research into other areas of chemistry and new collaborations are welcome.
[click here for recent publications]
Research is rarely only the result of one individual’s effort or inspiration. I would like to acknowledge collaborations in the Computational Chemistry group with Prof. Ian Hillier and Dr Mark Vincent who both have made significant contributions to these research highlights. We have been particularly lucky to have many very talented students and postdoctoral researchers in the group and it is to their credit that many have developed careers in computational research. These include postdoctoral positions in Germany, Belgium, USA and UK or in industries such as GSK, AstraZeneca, Tripos and BioFocus, and of course the few renegades who have preferred to bask in the Green lights of the City. I would like acknowledge their efforts and also those of our other academic co-authors over recent years, particularly to Dr R. Bryce, Dr J. McNamara, Dr C.I.F. Watt, Prof. A. Masters, Dr J. McDouall, Prof. M. Sutcliffe and Prof. N. Scrutton, all who feature in the selected references.
If you are interested in joining our research group please contact Dr Neil Burton.
We would also like to thank the NW-Grid Consortium and EPSRC for funding and computational resources.